4月22日から高橋輝行医師の外来のWEB予約を開始します。
事前にWEB上で公開されている問診票にご記入頂くとスムーズです。
予約優先ですが、従来通り予約なしの直接来院の患者様も順番に拝見致します。
是非、WEB予約をご活用下さい。
医師二人体制によるスムーズな患者様対応

20年以上の経験を有する専門医による診断と治療

地域に密着したクリニックとして幅広い科目に対応

お子様からご高齢の方まで様々な世代の患者様に対応

充実した医療機器(オープンタイプMRI装置・超音波診断装置)

予防接種や健康診断・人間ドックなどの自由診療にも積極的に対応

予防接種や健康診断・人間ドックなどの自由診療にも積極的に対応

診療カレンダー
(理事長)
| 日 | 月 | 火 | 水 | 木 | 金 | 土 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
| ||||||||||||||||||||
| ||||||||||||||||||||
| ||||||||||||||||||||
| ||||||||||||||||||||
| 日 | 月 | 火 | 水 | 木 | 金 | 土 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
| ||||||||||||||||||||
| ||||||||||||||||||||
| ||||||||||||||||||||
| ||||||||||||||||||||
午前(通常診療)・午後(認知症・頭痛・めまい・痺れ専門外来)
午前(通常診療)・午後(訪問診療(往診))
祝日
日野市立病院外来
午後休診
終日休診
休日診療(9時~17時)
17時 (受付は16時45分) 終了
診療カレンダー
(院長)
| 日 | 月 | 火 | 水 | 木 | 金 | 土 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
| ||||||||||||||||||||
| ||||||||||||||||||||
| ||||||||||||||||||||
| ||||||||||||||||||||
| 日 | 月 | 火 | 水 | 木 | 金 | 土 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
| ||||||||||||||||||||
| ||||||||||||||||||||
| ||||||||||||||||||||
| ||||||||||||||||||||
午前休診
午後休診
終日休診
受付時間変更
休日診療(9時~17時)
祝日
大熊医師(非常勤)の代診(9時半~12時)
大熊医師(非常勤)の代診(15時~17時)
友だち追加
 |
@rye5250j LINEの「友だち追加」から「ID検索」 または「QRコード」で登録 |
青和クリニック外来医師の
WEB予約はこちらから
 |
混雑緩和のため、予約優先となります。 直接来院された場合、混雑状況ではすこしお待たせすることもございます。 また、ワクチン接種もweb予約が可能です。ぜひご活用ください。 https://medicalpass.jp/hospitals/seiwa |
お知らせ
4月から当院でのBCGワクチン接種が開始となります。
また、6月から乳児健診(6,7か月健診と9,10か月健診)を再開します。予約は当院までお問い合わせください。
ワクチン接種で来院された際などに受付にお声掛けください。
※BCGワクチンの予約はwebには対応しておりません。(すでにお問い合わせいただいた方は申し訳ありません。)
5月以降の循環器科・腎臓内科受診について
5月以降に循環器科・腎臓内科を受診希望の方は、月曜、火曜午前、金曜、土曜日の受診をお願いいたします。
5月より、火曜午後の一部、水曜日午前、木曜日午前の担当医が大熊医師(非常勤)となります。詳しくはカレンダーをご覧ください。
※大熊医師の午前の診察時間は9時半からとなります。
循環器内科、腎臓内科の診察を希望される方は、月・金・土曜日での予約をお願いいたします。
高橋佐智子医師の外来は、土曜日以外も4月より完全予約制に移行します。
どうぞweb予約をご活用ください。
4月1日より生後2か月のお子様からから5種混合ワクチンの接種が可能になります。
今まで4種混合で接種をされていたお子さんは、引き続き4種混合ワクチンでの予約をお願いします。
◆4月の予定をお知らせ
4月16日(火)午後は、院長・理事長とも休診となります。
木曜日は引き続き休診となっています。
先日まで新規駐輪場の設置工事ならびに、駐車場の拡張工事を実施し、先日完了しました。
新規駐輪場は写真の位置になります。
車止めはなく、屋根の設置をしてあります。この場所への車の駐車は危険なため、おやめください。
引き続き駐車場を利用する際には、歩行者、運転者ともお気を付けください。
また、新規の駐車場もありますので混雑の際はご利用ください。

土曜日は、混雑緩和のため当面予約の方のみの診療といたします。
12月より当院で実施の乳児検診を休止します。
10月10日(火)より、65歳以上の方のインフルエンザワクチン接種が始まります。受診の際に医師にご相談ください。
一般の方は、10月23日(月)より接種を開始します。予約は、webのインフルエンザ
ワクチンの予約ページよりお願いします。
(院長診察または予防接種のページとは別になっています。かならずそこからの予約をお願いします。)
予約の開始は10月10日(火)からとなりますので、予約サイトをご覧ください。
https://medicalpass.jp/hospitals/seiwa
問診票はダウンロードしていただき、自宅で事前に準備してから来院をお願いします。
問診票のダウンロードはこちらへ
9月より、webにて予約が取れるようになります。日野市在住で50歳以上の方を対象として、不活化ワクチンの接種をご希望の方の予約を受付いたします。
https://medicalpass.jp/hospitals/seiwa
帯状疱疹の予約ページがありますので、そちらよりご予約をお願いいたしいます。
詳しくはスタッフ募集ページを御参照下さい。
令和5年8月18日から金曜日の外来は17:00 (受付は16:45) で終了とします。
17:00以降の外来受診を希望される方は月曜日か水曜日にお越し下さい。
送迎サービスは令和5年7月をもって終了致しました。
令和5年5月8日に新型コロナウィルス感染症が5類感染症に移行しました。
法令に則り、当院の発熱外来は終了致しました。
今後、発熱或いは風邪症状の患者様は引き続き一般外来で院内で診療致します。
5月よりweb予約を開始しておりますが、すでに多くの方に利用していただきありがとうございます。
現在発熱症状のお子さんも多く、予約せず直接来院の方が大変多くいらっしゃいます。
つきましては、外来での混雑緩和のため予約なしの方の受付は、10時半までとさせていただきます。
引き続き、予約の方優先で診察を進めさせていただきます。
また、web予約をしたが予定を変更し来院されない場合には、かならずキャンセルの操作をお願いいたします。
詳しくはスタッフ募集ページを御参照下さい。
5月8日から、院長(女性医師)外来のみwebでの予約が可能になります。
混雑緩和のため、予約優先となります。
直接来院された場合、混雑状況ではすこしお待たせすることもございます。
また、ワクチン接種もweb予約が可能です。ぜひご活用ください。
https://medicalpass.jp/hospitals/seiwa

5月8日からCovid19感染症の分類が5類に移行することなりました。
引き続き発熱の方の診察は行いますが、今までのように駐車場で外で待つことはなくなります。
当面の間、木曜日は休診といたします。
~痛みは「我慢」より「コントロール」を~
日常生活に支障を来すひどい生理痛を月経困難症といいますが、思春期ではそのほとんどが、子宮が未成熟であることに起因します。痛みに対して鎮痛剤を使用する場合、我慢の限界を待つより、痛みそうと思ったらすぐに飲む方が効果的です。また、一部に子宮内膜症が隠れている場合があり、進行すると将来の妊娠に影響する可能性があります。子宮内膜症の予防と症状の軽減には低用量ピルが有効で、世界的にも初経以降は使用が可能とされています。生理痛は誰にでもあるものと我慢させず、ご相談ください。(内診は行いません。低用量ピルは月経困難症の保険適応があります。)
※成人ですでに月経困難症で診断・治療を受けている方は、基本的にはかかりつけの産婦人科を受診してください。
3月より、当面の間木曜日は休診といたします。
JCB・AMEX・VIZA・マスター・Diners のカード決済が可能です。
ただし、1万円未満のカード決済はお断り致します。
患者様が診察の順番待ちをされる際に、当院の駐車場に停めたご自分の車でお待ちいただける設備「コールベル」を準備いたしました。
ご希望の方は受付の際にお申し出ください。診察の順番がきましたら、音、振動、光でお知らせいたします。それまで当院の駐車場に車を止めていただき車内で待っていただくことが可能です。コールベルは、お知らせの後受付にご返却ください。

昨今の新型コロナウイルス患者の発生状況から、当院での6月から初診の電話でのオンライン診療は中止とさせていただきます。
また、かかりつけの方の再診の場合も電話でのお話のやりとりではなく、可能な限り受診していただき診察を受けられての処方が望ましいと考えます。
基本的に患者様の症状やその後の経過は、対面診療が大切であり慢性疾患の方でも自覚症状がすべてではありません。
診察室では、触診や聴診以外にも患者様の顔色や表情、歩き方など総合的に診察をしています。
どうしても、お電話で「変わりありません」というお話だけでは、診療に必要情報は不十分となります。
待ち時間が気になる場合には、事前の予約をしていただきますと比較的スムーズにご案内できると思われます。
また、ご自身の症状について受診してもよいかどうかご心配な場合にもお電話をしていただいて構いません。
どうしても受診できないという事情がなければ、ぜひクリニックに足を運んでいただきお顔を見せていただければ、皆様により良い医療が提供できると考えます。
当院の方針につき、ご理解をお願いいたします。
院内での様々な感染症の拡大を予防するため、患者様には受診される際にご自宅よりマスクの着用をお願いいたします。
万が一お持ちでない場合には、受付にてマスクをご購入いただきます。購入は受診される方と付き添いの方の分のみとなります。
10月からの消費税の変更に伴い、厚生労働省が定める診療報酬や薬価が一部引き上げられることになりました。 そのため、他の医療機関同様に保険診療ならびに自費診療における患者様の窓口でのお支払いが、9月以前より変わる場合がございます。 ご理解のほどよろしくお願いいたします。
ポータルサイト「睡眠時無呼吸なおそう.com」のご覧になってください。当院も登録されています。
睡眠時無呼吸外来を新設しました。詳しくは「睡眠時無呼吸症候群外来」のページを御参照ください。「私の体験談」を掲載したのでぜひご覧になってください。
妊娠中の方、授乳中の方で婦人科症状以外の風邪症状などの体調の悪い方は、当院は診察可能です。
◆妊娠中の方、授乳中の方で婦人科症状以外の風邪症状などの体調の悪い方は、当院は診察可能です。
お気軽にご相談ください。診察の際に必ずその旨をお伝えください。
◆当クリニックのラインアカウントができました。
休診情報などをお知らせしますので、QRコードまたはIDから登録してください。
当院のMRI脳ドックをリニューアルし、内容と価格設定を一新しました。
◆当院のMRI脳ドックをリニューアルし、内容と価格設定を一新しました。
詳しくは「MRI脳ドック」のページを御参照ください。
なお、「MRI脳ドック」は原則、水・金曜日の午後と土曜日の午前に実施します。
脳ドックの予約サイトはこちらです。
詳しくはクリニックまでお問い合わせください。
理事長外来に関して、新たに月水金の午後に「認知症・頭痛・しびれ・めまい専門外来」を開設致します。
◆理事長外来に関して、新たに月水金の午後に「認知症・頭痛・しびれ・めまい専門外来」を開設致します。
◆診察時間変更のお知らせ
2017年9月19日から午後の診察時間の開始を原則15:00~と致します。
◆院長午後休診のお知らせ
院長の水曜日の午後の外来は休診となります。
◆リハビリ情報局を更新しました。
御興味のある方はぜひ御覧になってください。
リハビリ情報局のページはこちら
当院の特徴





診療案内






医師紹介
理事長 高橋 輝行(たかはし てるゆき)
医療法人社団 桃實会 青和クリニック理事長の高橋輝行です。
当院の理念は、医療人である自分やその家族、友人が安心して通院できるクリニックを目指し、患者様の立場に立ち、患者様に信頼され納得していただける良質な医療サービスを提供することです。
加えて、高齢化社会を担うに十分な経験と設備を兼ね備え、ご高齢の患者様や障害のある患者様が安心して受診できるクリニックを目指しています。
医師紹介
院長 高橋(旧姓 野中) 佐智子(たかはし さちこ)
はじめまして。医療法人社団 桃實会 青和クリニック院長に就任いたしました、高橋佐智子です。
多くの方々のご協力により、生まれ育ったこの地でクリニックを開院することができました。
卒業した日本大学医学部附属病院である板橋病院では、循環器・腎臓・内分泌内科に所属し、医療に携わっておりました。
日本大学板橋病院の良いところは、専門科の疾患だけではなく、内科全般について学べるところです。私は、様々な疾患の救急対応などもできるようなトレーニングを受けてまいりました。
大学院では、腎臓内科学を専攻し、慢性腎炎のもっとも多い原因の一つである、IgA腎症の研究で、学位を取得いたしました。大学院で研究をしている間に、第一子を出産し、育児と仕事を両立させてきました。
その他、検診担当医や産業医を経験し、生活習慣病に関する食事指導や運動指導にも積極的に取り組んでまいりました。また、平成20年からは日野市立病院の内科と健診センターで、多くの地域の患者様の診療にあたってまいりました。
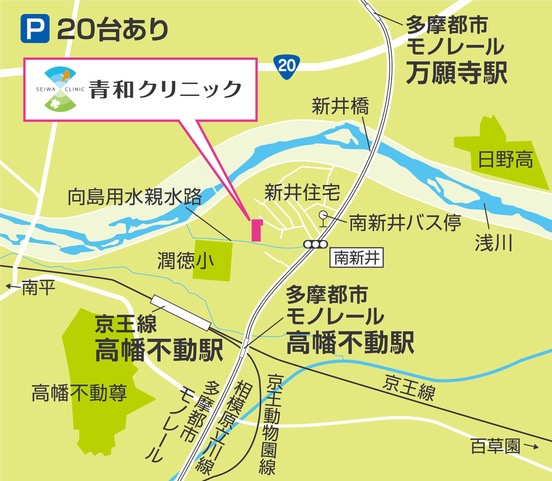
医院案内
青和クリニック
【理事長】 高橋 輝行
【科目】内科、神経内科、小児科、リハビリテーション科
| 診療時間 | 月 | 火 | 水 | 木 | 金 | 土 | 日祝 |
|---|---|---|---|---|---|---|---|
| 9:00~12:00 受付時間 8:45~11:30 |
● | ● | ● | ▲ | ● | ● | / |
| 15:00~19:30 受付時間 14:45~19:00 |
◆ | ● | ◆ | ▲ | ■ | / | / |
●…訪問診療(往診)
▲…日野市立病院外来
◆…認知症・頭痛・めまい・痺れ専門外来
■…令和5年8月18日より、15:00~17:00 受付時間 14:45~16:45
日曜日と祝日は休診
【院長】 高橋 佐智子
【科目】内科、循環器内科、腎臓内科、小児科
| 診療時間 | 月 | 火 | 水 | 木 | 金 | 土 | 日祝 |
|---|---|---|---|---|---|---|---|
| 9:00~12:00 受付時間 8:45~11:30 |
● | ● | ● | / | ● | ● | / |
| 15:00~17:00 受付時間 14:45~16:45 |
● | ● | / | / | ● | / | / |
日曜日と祝日は休診
MRI 脳ドック(完全予約制)
| 診療時間 | 月 | 火 | 水 | 木 | 金 | 土 | 日祝 |
|---|---|---|---|---|---|---|---|
| 9:00~12:00 | / | / | / | / | / | ● | / |
| 15:00~18:00 | / | / | ● | / | ● | / | / |
診察は理事長の高橋輝行が担当します。
日曜日と祝日は休診
@rye5250j
LINEの「友だち追加」から
「ID検索」または「QRコード」で登録
青和クリニックからの情報を発信します。
個人情報の送信にはご注意ください。